Images of リスクマトリックス







6 Questions for Origami Risk’s Chris Bennett on Risk Management Tech AdvancementsIn Today’s Polarized Culture, Companies Must Have Clear Policies for Employee Speech to Avoid Reputational Damage and EPL Claims11 Ways Inflation and Economic Volatility are Influencing Insurance in 2023

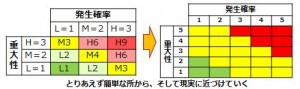

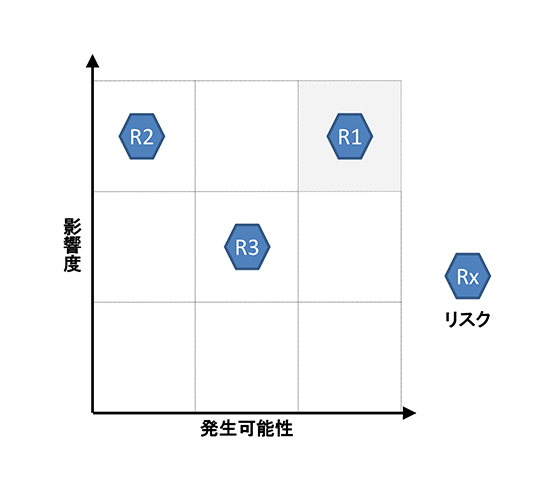

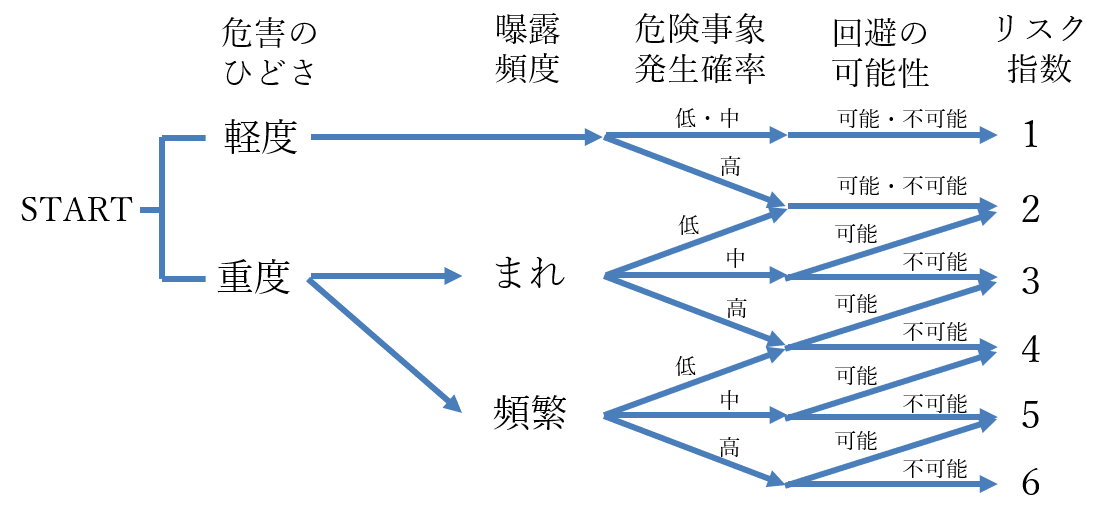

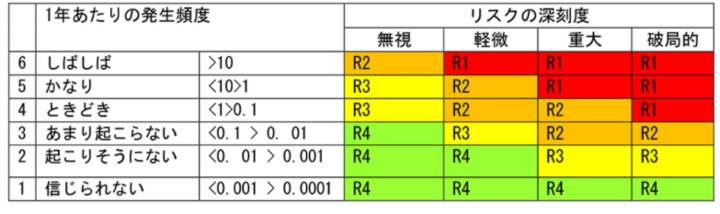

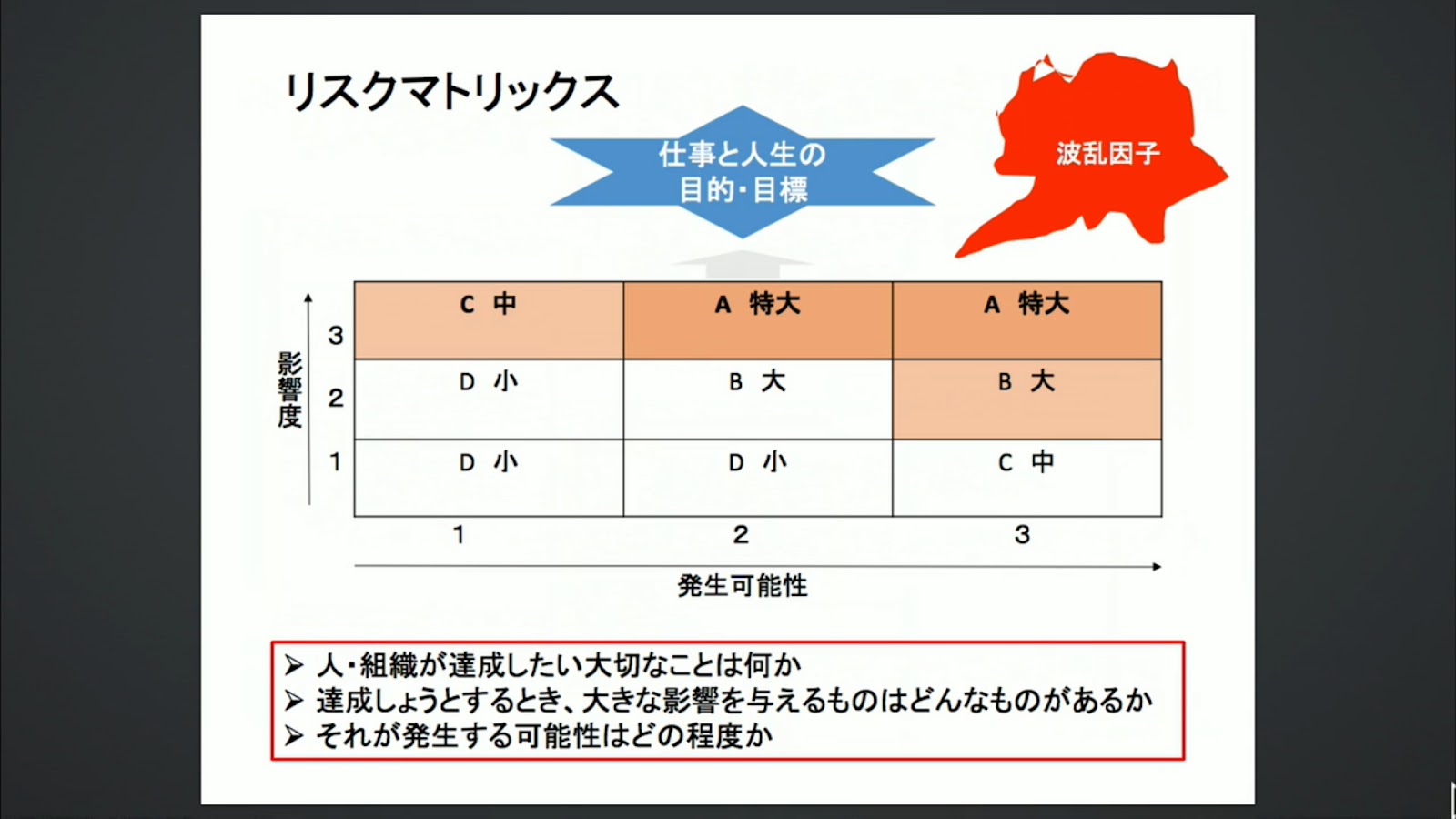
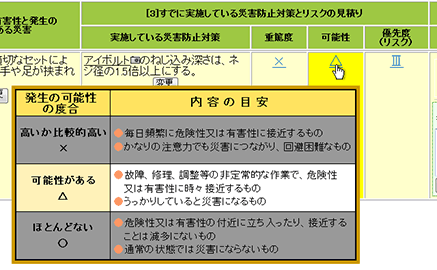


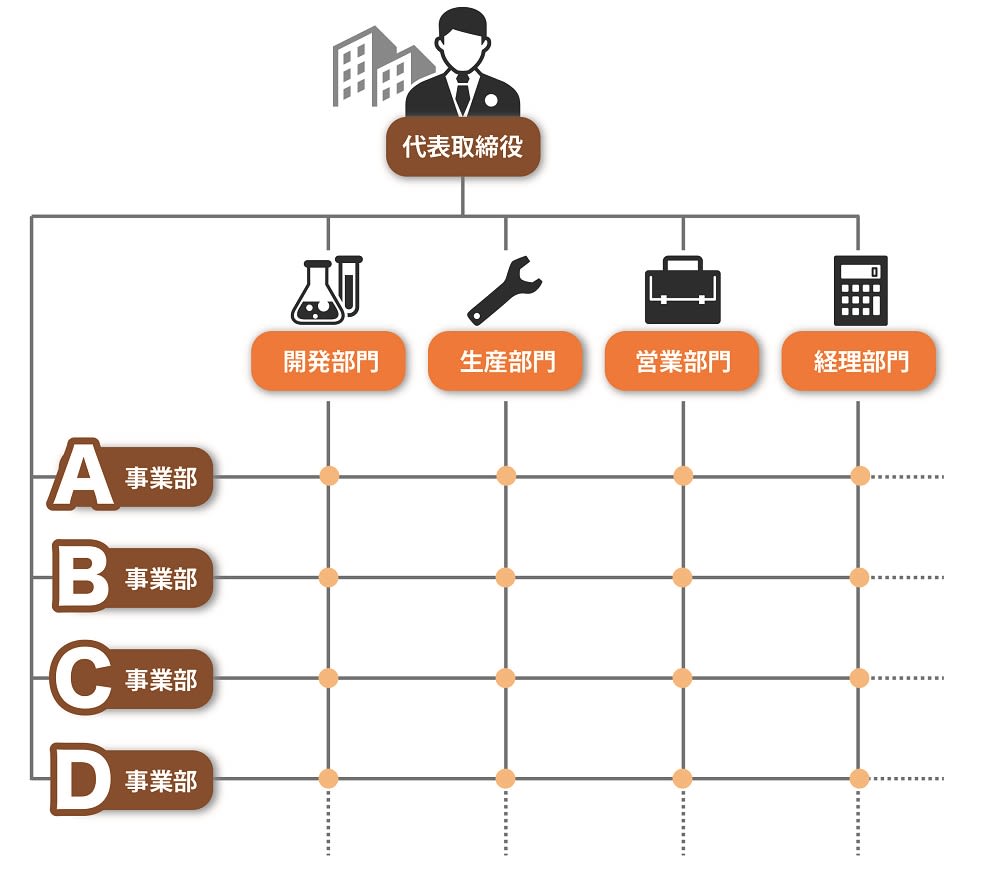

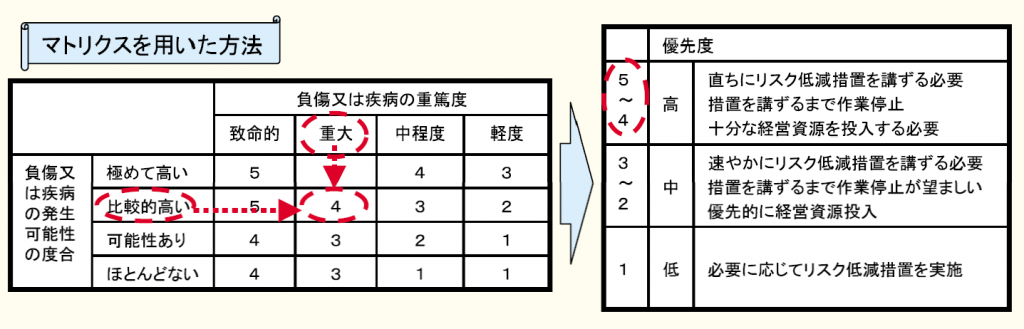



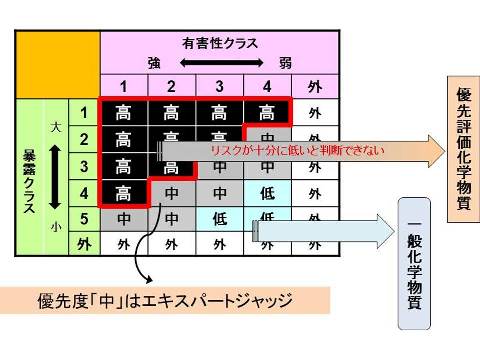
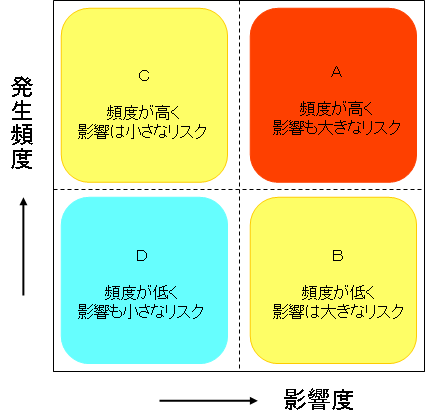
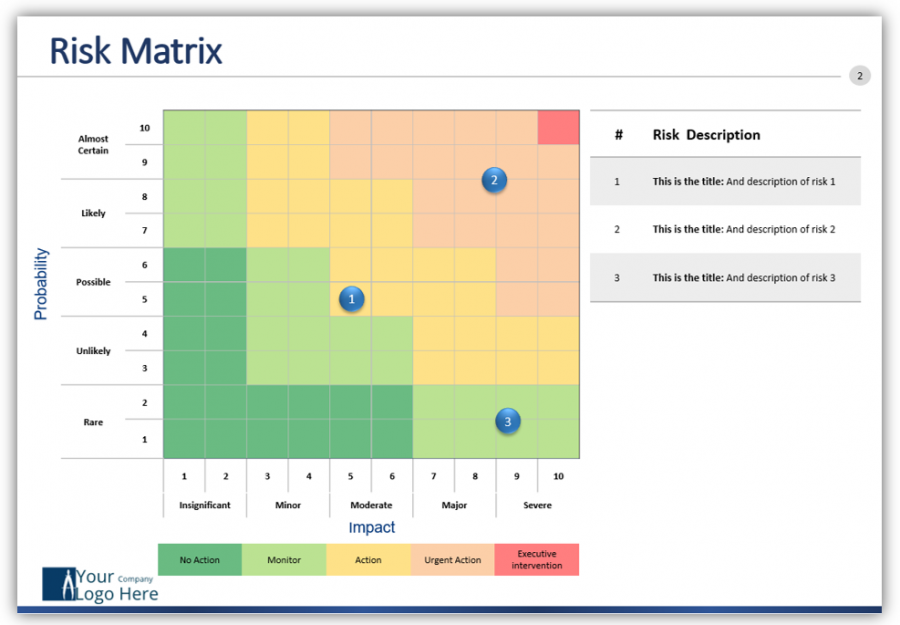
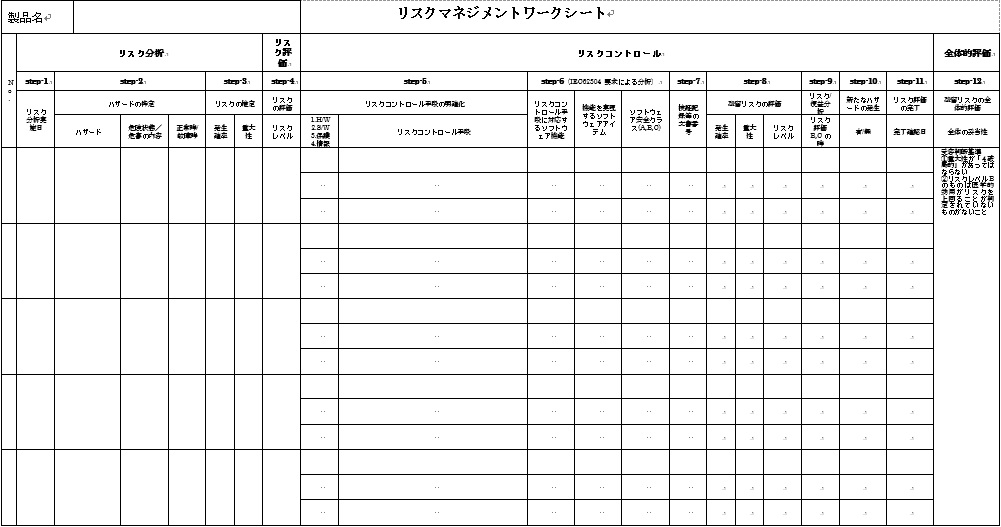
医療機器企業におけるリスクマネジメントについて研究するページ1. はじめに 医療機器には何らかのリスク(危害の潜在的な源)が存在している。何らかのリスクを抱えた状態で市場 へ出荷され、事故が起こる前に予防策として、あらゆるリスクを検出し、それによって患者やユーザに危害 を及ぼさないようにするのが、医療機器におけるリスクマネジメントの主目的である。 製品実現の中でリスクマネジメント活動を行い、特に製品の設計・開発にあたっては、リスクマネジメン トから得られた情報を加えることが必要である。つまり、市場からの製品(類似製品を含む)におけるフィ ードバック情報も重要な要素となる。 この活動はPDCAを基本としている。必ず市場からのサーベイ、顧客からの苦情、そういうものをフィー ドバックしてリスク分析に戻さないといけないのである。ここでいうリスクとは、「患者やユーザに対する健 康被害」のことである。「医療機器に欠陥が潜んでしまう」や「製造担当者がけがをする」といった事象は、 リスクマネジメントの対象ではないことに注意が必要である。 1つの医療機器の設計の全ライフサイクルを通じて、医療機器企業がリスクコントロールを最大限実施し たことを証明するために、リスクマネジメントにおける活動は、リスクマネジメントファイルにおいて、完 全に文書化しなければならない。 適切なリスクマネジメント活動は、新しい医療機器の開発、生産、販売等を支えることにより、重要な価 値をもたらす。 リスクマネジメント活動により、製品開発スケジュールへの影響を最小限に留めながら、機能的安全性や 使い勝手を向上させるための変更や改良の特定や実施が可能になる。 また、製品を市場に投入するまでの時間を短縮して、一層の競争優位性をもたらす。 2. 品質が良い医療機器とは 品質が良いということは、どんな状態であろうか。 医療機器においては、ユーザの要求事項と完成した医療機器の仕様が完全に適合している状態が、品質が 良いといえる。 使い勝手が良いとか、見てくれがいいとか、デザイン性がいいとか、そういうことは無関係なのである。 医療機器は、意図される使用目的(intended use)に対して機器が完全に適合している状態、これが、品質 が良い医療機器なのである。 要求事項と完成医療機器が完全に整合していることを確認して保証することを「バリデーション」と呼 ぶ。日本語では「妥当性の確認」と訳されている。 ISO-9000による定義には、「バリデーションとは、特定の意図した用途またはアプリケーションに関する要 求が満たされていることを、客観的な証拠の提示により確認することである。」と記載がる。 また、QSRには、「バリデーションとは、特定の意図する用途のための特有の要求事項が恒常的に満たされ 得るという客観的証拠の検討と規格による確認を意味する。」と記載されている。 バリデーションは、必ずユーザ環境またはユーザ環境を模擬した環境で実施しなければならない。医療機 器企業側のみで実施すると、実際の使用状況と異なり、想定外の事故が起きてしまう可能性があるのである。 ところが、医療機器には1つ問題がある。そもそも要求事項が正しいとは限らないのである。例えば、医 師などから、新規医療機器に対する要求が得られたとしよう。その要求に従い、設計し、製造したとしよう。 しかしながら、実際に使用してみた場合、実は当初想定したような使用目的にそぐわず、使いづらいなどの 問題がしばしば発生する。そのため、何らかのリスクが潜んでしまったり、時には事故が発生してしまうのである。 品質には4つの種類がある。 1. 企画の品質 2. 設計の品質 3. 製造の品質 4. サービスの品質 である。 それぞれ、品質の4つの種類における要求事項は、次の通りである。 企画の品質においては、製品で実現しようとしている特性に対する顧客の要求である。 設計の品質においては、企画の段階で検討された特性の水準や品質仕様である。 製造の品質においては、図面・仕様書などの設計文書である。 サービスの品質においては、調整、据え付け、消耗品の補給、不良品などへの対応に対する顧客の要求で ある。 これら要求事項を満たした場合、当該医療機器および付帯サービスは、品質が良いといえるのである。 3. ISO-13485の認証とFDA査察の違い ISO-13485は、「医療機器の品質マネジメントシステム−規制目的のための要求事項」である。 ISO-9001は、製造業からサービス業まであらゆる業種に対応可能な反面、医療機器に適用しようとす ると、その安全性を確保しきれないという問題があった。そこで制定されたのが、ISO-13485である。 ISO-9001をベースに、医療機器の安全性や品質を確保するのに必要な要求事項が追加された。 ISOに参加する各国は、国民の健康維持・向上のため、医療機器を厳しく規制している。ISO-13485が 「規制目的のための要求事項」とされているのは、このためである。 ISO-13485の目的は、世界中の医療機器法令の整合化を促進することである。つまり、ISO-9000は民 間の規格であるのに対して、ISO-13485はもはや規制要件なのである。 日本、米国、欧州など、ISOに参加する各国の当局がレビュを行っている。 ところで、ISO-13485の認証を受けていても、FDA査察で指摘されることがしばしばある。 ISO-13485の認証は、第三者認証機関(NB:Notified Body)が実施する。つまり民間業者がビジネス として実施するのである。クライアントとの付き合いもあり、必然的に厳しい指摘には及ばないことや 多くの指摘を出さないこともしばしばである。 しかしながら、FDAは米国民を保護するために、日本までやってきて査察を行うのであるから、リス クを徹底的に調査し、指摘を出すのである。査察官自身も国民への説明責任があり、また事故などが発生 した場合には、査察した者の責任が問われることもあり得るのである。 4. 放射線治療装置の死亡事故 1985年から1987年にかけて、米国で複数の医療施設でTherac-25という放射線治療装置が誤作動し、過大 な放射線を浴びた患者に死傷者が出た。 Therac-25という放射線治療装置は、旧モデルのTherac-20の廉価版であった。そのコストの下げ方は、電 気機械式の安全保護装置を、「安全はソフトウェアでも確立できる」というコンセプトのもと、経済性を優先 させるためにハードウェアによる安全装置を外した。そのためハードウェアによる安全装置のために抑えら れていたソフトウェアの不具合が発現した。 問題は、医療機器企業でのテストは、テストスクリプトに従ってテストデータを入力し、確認をしながら ステップを踏む。しかしながら、実際の医療現場では、医師がいわゆるブラインドタッチを行い、素早くデ ータを打ち込むことが多い。そのため、高エネルギーの放射線を照射してしまったために、被爆した患者が 数日後に亡くなったということであった。Therac-20では、ハードウェアによる安全装置が存在したため、当 該事故は発生せず、ソフトウェアの問題は露見しなかったのである。 当初は、調査したFDAも当該医療機器企業も、ハードウェアの故障と考えていた。そのため、なぜ過度な エネルギー照射が起こるのか原因を突き止めるのに長い時間がかかり、付け焼き刃の対策で装置の使用を止 めなかったために、Therac-25による犠牲者は6名にまで拡大してしまった。 この事故を調査した現マサチューセッツ工科大学のNancy Leveson教授は、事故調査を終えて次のような 教訓を書き残している。 ・ソフトウェアはどんなに慎重に作っても満足できるほどに完璧にはならないため、ソフトウェアがどん な動きをしたときでも、人命に危険が及ばないようにシステムを設計する必要がある(フェールセーフ、 独立した安全装置の設置など) ・ソフトウェアの信頼性をできるだけ高めるのは悪いことではないが、それでソフトウェアが安全になる わけではない。プログラマは要求を満たすようにコードを書くが、要求が正しいとは限らない。事故の ほとんどは、プログラミングのエラーではなく、要求が間違っていたり、不完全であったりしたために 起きている ・われわれの目標は、だれもがこの経験を生かせるようにすることであり、装置のメーカーや他の人を批 判することではない。間違いは、このメーカーにだけ起きたのではなく、残念ながら、安全が最優先さ れるべきその他のシステムにも、きわめて日常的に起きている ・いくつかの出来事が複雑に絡み合って起きた事故が、1つの原因にされることが多すぎる。ほとんどの 事故は、システム事故、つまり、さまざまな構成部分や機能が複雑に作用しあって起きるものだ。事故 の原因を1つに特定することは、大きな間違いだ。 Nancy Leveson 教授が語ったTherac-25の事故調査から導き出された組込みソフトウェアの安全に関する教 訓は、20年以上経過した現在でも生き続けている。 というよりも、組込みソフトウェアの大規模・複雑化とCPUの統合化により、より危険性が加速されてき おり、組込みソフトエンジニアがソフトウェア開発を行っている際に常に考えていなければいけない重要な 教訓となっている。 この事故を教訓に、FDAは1987年に「General Principles of Software Validation(ソフトウェアバリデーショ ンの原則)」と呼ばれるガイドラインを発行した。 ソフトウェアのバリテーションは、医療機器メーカーだけではなく、ユーザの環境またはユーザ環境を模 擬した環境で、ユーザが参画して実施することが義務付けられたのである。5. 誤使用 5.1 合理的に予見可能な誤使用 市場出荷された医療機器自体になんら欠陥がないとしよう。ところが、使う側であるユーザが操作を間違ってしまうということがあるのである。ヒューマンエラー、誤使用、不注意というものに対しても、必ず発生するので生産者側では気をつけなくてはならない。 医療機器の場合においても、こういった誤使用という問題がある。医療機器業者が想定した使用法が記載 されている取扱説明書の通りに使用しないというのが誤使用である。これはヒューマンエラーである。 いくら医療機器企業側が気をつけていても、ユーザ側が間違ってしまうということも、リスクとして捉え ていないといけないのである。 ISO/IEC Guide 51では、設計者等が想定した使用法(intended use)のみならず、「合理的に予見可能な誤使 用」(reasonably foreseeable misuse)も含んだ範囲でリスクを評価し、受け入れ可能なレベルに達するまで、リ スク低減を行う反復プロセスを図示している。(図2参照) 「合理的に予見可能な誤使用」をリスクアセスメントの範囲とすることを明記したことが極めて重要である。 5.2 誤使用の例 医療機器以外での誤使用、不注意に分類される事故例としては、ガスコンロの消し忘れで火がついてしまった、少ない油で天ぷらを揚げて火災になった、換気扇を回さずにガス湯沸かし器を使って一酸化炭素中毒になってしまったなどがあり得る。 リビングや寝室では、石油ストーブに洗濯物が落下して火災が発生した、付近の可燃物が発火した。湯た んぽでは低温やけどが発生した、電池を逆に入れてしまって破裂した、間違ってボタン電池を飲み込んでし まったなどが実際に起きている。 このようなことを含め、生産者はあらたな誤使用というリスクを発見した場合、そのリスクを回避するよ うな設計に変更にしなければならないのである。 または、誤使用を未然に防止するよう、ラベルなどに注意書きを記載しておかなければならない。例え ば、レーザーポインターには、「直接、目でのぞかないでください。」と注意書きが貼ってある。または携帯 用のカイロでは「肌に直接貼らないで下さい。低温やけどの恐れがあります。」と記載がある。 それでも、人は必ず間違いを起こすのである。 1980年の日航ジャンボ機の墜落事故は、ボーイング社の修理ミスだといわれている。医療事故では輸血す るときに血液型を間違えたとか、患者を取り違えたとか、薬品を間違った、手術のときに左右の肺を間違え た、そういったこともしばしば発生している。 株の大量誤発注という事件もあった。61万株を1株1円で売りに出してしまった、数量と金額を逆に間違 えて入力してしまったのが原因である。その際、東証のコンピュータには、発注取り消しという機能が設計 されておらず、被害が拡大した。 6. ISO-14971 6.1 ISO-14971とは ISO-14971「医療機器 - リスクマネジメントの医療機器への適用」は、医療機器企業が体外診断用医療機 器を含む医療機器に関連するハザードを特定し、リスクの推定および評価を行い、これらのリスクをコント ロールし、そのコントロールの有効性を監視する手順について規定しているというものである。 ISO-14971は、リスクマネジメントの原則と実践について示したものである。 ISO-14971は、医療機器に関する幾つかの主要な規格において参照されている。例えば、ISO-13485(医療 機器の品質マネジメントシステム)、IEC 60601-1(電気安全性)、IEC/EN 62366(医療機器の使い易さ)、ISO 10993(生物学的評価)、IEC62304(医療機器のソフトウェア)などである。 ISO-14971で示されたリスクマネジメントのプロセスは、次のような内容となっている。 6.2 ISO-14971の適用範囲 ISO-14971は、組織の営業活動から設計開発、製品製造、検査測定、製品納入、付帯サービスおよび市販後 の調査に至るすべてのライフサイクル活動に適用される。 しかしながら、大事なのは、医療機器の「設計・開発」プロセスにおいて、リスクマネジメント活動の結 果を取り入れて活動しなければならないということである。医療機器は医薬品と異なり、製造工程よりはむ しろ設計工程でリスクマネジメントを主に実施しなければならない。 設計・開発が適用除外される医療機器であっても、製品実現全体をとおしてリスクマネジメントのための 文書化された要求事項を確立することが要求されている。 リスクマネジメントの活動対象範囲は、組織が製造する製品すべてが対象となる。少量多品種であっても その一部を適用除外することは出来ない。また、付属品等も適用対象になる。 ただしISO-14971は、臨床的判断には適用しない。また、受容可能なリスクレベルを規定していない。 6.3 ISO-14971の経緯 ISO-14971の経緯を紹介する。(図3.参照)6.4 ISO-14971の構成 ISO-14971の目次を図4に記載する。 6.5 IEC-60601-1とISO-14971 IEC-60601-1には、「ISO-14971に適合するリスクマネジメントプロセスを実施する。」という要求事項があ る。また、要求事項の適合性の確認について、「適合性は、リスクマネジメントファイルを調査して確認す る」と記載がある。 しかし、IEC-60601-1の要求事項に適合していれば、その要求事項についてはリスクマネジメントを実施し なくても良いという「基礎安全」の考え方を取り入れている。 つまり、機器全体はリスクマネジメントISO-14971の管理下にあるが、その中でIEC60601-1の要求事項に よる部分があるということである。 IEC-60601-1の要求事項では、副通則であるEMC(IEC60601-1-2)、alarm(IEC60601-1-8)、usability (IEC60601-1-6)も含まれている。 7. EN ISO-14971 2012年7月31日、EN ISO-14971: 2009は、EN ISO-14971:2012に置き換わり、2009年版は2012年8月30 日の時点で廃止された。 EU地域では、EN ISO-14971:2012は、2013年1月より強制になった。EU圏へ輸出する製品への影響を評 価する必要がある。各国・地域の対応は、その国・地域の事情もあって独自路線をとるのはいたしかたない ところである。 EN ISO-14971:2012は、ISO-14971:2007の規定についての相違はないが、informative(参考)である附属書 Zs(Za、Zb、Zc)が変更されている。3つのEU医療機器指令(MDD:Medical Device Directive)の基本要件 (ER:Essential Requirements)の一部を加えたものになっている。 EN ISO-14971:2012は、以下の7つの点において、ISO-14971:2007と異なっている。 7.1.1 “negligible risk”(無視可能なリスク)の扱い “negligible risk”(無視可能なリスク)の扱いとして、ISO-14971の定義では“negligible risk”(無視可能な リスク)を放棄することができる場合があると記載されている。 EN ISO-14971では、全てのリスクについて、リスクマネジメントを実施しなければならないと記載され た。 7.1.2 リスクの受容可能性についての医療機器企業の自由裁量権 ISO-14971では、リスクの受容可能性の基準は医療機器企業が決定することになっている。その上で、受容 不可能なリスクに対してのみ、それらを統合した全体のリスク/便益分析を行うこととしている。 EN ISO-14971では、医療機器企業がリスクの受容可能性を判定するのではなく、全てのリスクは可能な限 り低減されなければならないとしている。 EN ISO-14971では、もはや受容可能性という概念はないに等しく、全てのリスクは他のリスクと組み合わ せてリスク/便益分析を実施しなければならない。 7.1.3 ALARPではなくAFAPによるリスク低減 ALARP(as low as reasonably possible:合理的に実行可能まで低く)ではなく、AFAP(as far as possible:可 能な限り)によるリスク低減を要求している。 ISO-14971では、リスクは「合理的に実行可能な程度まで(ALARP)」低減することと記載されてい るが、これは経済性を考慮した要求である。あまりにも規制を厳しくすると、コンプライアンスコストが 上昇し、製品の価格に上乗せされ、結果的には患者負担になってしまう。 EN ISO-14971では、「可能な限り(AFAP)」低減することを要求しているのである。 リスクを徹底的に低減するということは、開発コストがかかってしまい、経済性を無視しているという ことである。 7.1.4 あらゆるリスクに対するリスク/便益分析の適用 ISO-14971では、受容可能性を超えるリスクに対して、リスク/便益分析を要求している。 EN ISO-14971では、個々のリスクおよびすべての残存リスクに対するリスク/便益分析を要求してい る。 7.1.5 リスクコントロール手段についての裁量 ISO-14971では、下記3つのオプションから1つを選んでリスクコントロールを実施することを要求 している。 7.1.6 初期リスクコントロール手段についての裁量 ISO-14971では、設計による固有の安全性を要求している。 これに対し、EN ISO-14971では、可能な限りリスクを低減させることを要求している。設計および材 料・組立による安全性を要求しており、範囲が広い。 7.1.7 残存リスクについてのユーザへの情報提供 ISO-14971では、残存リスクについてのユーザへの情報提供は、リスクコントロール手段の1つであ るとしている。取扱説明書に記載するというのも、リスクコントロールの1つなのである。 EN ISO-14971では、残存リスクについてのユーザへの情報提供はリスク低減手段ではないとしてい る。 8. リスクマネジメント手順 本章では、設計開発のインプットとなるリスク分析、リスクを低減するためのリスクコントロール、リス クの受容可能性の決定とリスク/便益分析、また製造中および製造後の情報によるリスクマネジメントの見直 しといった、医療機器のリスクマネジメント手順について解説をする。 リスクマネジメント手順は、以下のステップにより構成される。 8.1 意図する使用および医療機器の安全に関する特質/医療機器の使用に関する特性の明確化(Step-1) このステップでは、対象となる医療機器について、意図する使用および合理的に予見できる誤使用を明確化する。 必要な場合には、安全に影響する定性的および定量的特性または必要な場合には限界を明確にする。 ユーザビリティに関係するハザード抽出のため、 8.2 ハザードの特定(Step-2) このステップでは、当該医療機器に対する既知のハザードおよび予見できるハザードを特定する。(図6参照) 8.3 リスクの推定(Step-3) Step-2で特定した「危害」つまりリスクに対して、「発生確率」および「重大性」を推定する。 いずれも定量的な表現か、または定性的な表現が用いられる。(図7参照) 8.4 リスクの評価(Step-4) Step-3で求めた発生確率と重大性から、リスクレベルを算出する。 リスクレベル=重大性×発生確率であるので、図7に従って、A3、A2、A1、B3、B2、B1、Cのいずれかになる。 リスクマネジメントワークシートに記載する際には、A、B、Cのいずれかで良い。 8.5 リスクコントロール Step-4で求めたリスクレベルにより、リスクコントロールの要・不要(リスクの許容可能性)を判定する。 リスクレベルがB以上の危険状態については、以下を実施する。 8.5.1 リスクコントロール手段の明確化(Step-5) リスクレベルの低減のための手段(リスクコントロール手段)を明確化する。 リスクコントロール手段としては、次の優先順位に従い、それらの1つ以上を使用すること。 8.5.2 IEC-62304によるソフトウェアに関する分析(Step-6) ソフトウェアを搭載した医療機器(単体プログラムを含む)の場合、IEC-62304の要求に従って、ソフトウ ェアの設計を実施しなければならない。 一般にソフトウェアの設計では、ソフトウェア要求仕様書(Software Requirements Specifications:SRS)に 従って、アーキテクチャ設計書を作成する。 アーキテクチャ設計書は、ソフトウェア要求仕様書に対応するソフトウェアの機能を記述し、さらに各機 能をソフトウェアアイテムに分割する。ソフトウェアアーキテクチャ設計の終了は、開発における最も初期 のポイントである。ソフトウェアアイテムの明確化により、安全性との関連が特定可能になる。 IEC-62304では、アーキテクチャ設計において分割されたソフトウェアアイテム毎に、ソフトウェアアイテ ムが寄与因子であり得るハザードから生じる、患者、オペレータまたはその他の人々に対する考えられる影 響により、ソフトウェア安全クラスを割り当てることを要求している。 ソフトウェア安全クラスは、危害の程度に基づいて次のいずれかに分類する。 クラスA:傷害が発生しない クラスB:深刻な傷害が発生しない クラスC:死亡または深刻な傷害が発生するおそれがある ただし、ハザードから生じる危険が、ハードウェアのリスクコントロールにより受容可能なレベルに低減 できるのであれば、それらを考慮に入れても良いとしている。 8.5.3 リスクコントロール手段の実施と文書化(Step-7) 選択したリスクコントロール手段を実施する。つまり、ハードウェアやソフトウェアによる安全装置の設計や、取扱説明書やラベルなどに使用女王の注意などを記載する等である。 実施したリスクコントロール手段は、それぞれの設計書等に文書化すること。 また、リスクコントロール手段が適切で有効であることを検証した記録を作成すること。 8.6 残留リスク評価(Step-8) リスクコントロール実施後に残留するリスクレベルを評価する。リスクコントロール実施により減少した発生確率および重大性を求め、リスクレベルを算出する。 リスクレベル=重大性×発生確率 リスククラスが、図7においてCになっていれば、受容可能と判断する。 受容可能であると判断した残留リスクについては、どの情報を提供(開示)するか、かつ、どの附属文書に記載するかを決定すること。 リスクレベルが受容可能なまで低減されていない場合は、Step-5に戻り、リスクコントロール手段を明確化すること。 特定した全ての危険状態からのリスクが考慮されていること(リスクコントロールの完全性)を明確にすること。 8.7 リスク/便益分析(Step-9) Step-8において、受容可能でないと判断し、かつ、それ以上のリスクコントロールの実施が現実的でない場合、データおよび文献を収集し、かつ、見直した上で、意図する使用の医学的効用が残留リスクを上回るかどうか判断する。 重大性が「重大」、「中程度」「軽微」と判定されたリスクであっても、リスク/便益分析を実施すること。 ただし、重大性が「致命的」なリスクが残留した場合は、その製品の開発を打ち切らなければならない。 8.8 新たなハザードの発生確認(Step-10) リスクコントロール手段の実施によって、新たなハザードが発生し、影響が出ていないかを調査する。以 下を考慮すること。 8.9 リスク評価の完了(Step-11) 上記のすべてのステップを踏み、リスクマネジメント活動が適切かつ完全に文書化されていることを確認した上で、個々の危険状態に対する一連のリスク評価活動が終了する。 完了した日付を、リスクマネジメントワークシートに記載すること。 8.10 残留リスクの全体的評価(Step-12) すべての危険状態に対して、残留リスクの全体的な評価を行う。結論としては、当該機器における残留リスクが受容できるか、受容できないかのいずれかとなる。 ここで、重大性が「致命的」なリスクがあってはならない。 また、残留リスクのリスククラスがB以上のものに対して、医学的効用がリスクを上回るものであることの判定が、根拠をもって実施されていなければならない。 8.11 製造中および製造後の情報によるリスクマネジメントの見直し手順 製造中および製造後に「新たな問題」が発生していないかを調査する。新たな問題が発生している場合は、 リスクの再評価を行うこと。 次の検討を行う。 9. 体外診断用医療機器(IVD)のリスクマネジメントモデル 体外診断用医療機器(体外診断用医薬品を含む)は、患者に直接接触することはない。つまり、当該体外診断用医療機器の欠陥により、患者がけがをすることはない。そのため、リスクの評価があいまいになりがちである。 図8に、体外診断用医療機器のリスクマネジメントモデルを示す。

医療機器企業におけるリスクマネジメントについて研究するページ1. はじめに 医療機器には何らかのリスク(危害の潜在的な源)が存在している。何らかのリスクを抱えた状態で市場 へ出荷され、事故が起こる前に予防策として、あらゆるリスクを検出し、それによって患者やユーザに危害 を及ぼさないようにするのが、医療機器におけるリスクマネジメントの主目的である。 製品実現の中でリスクマネジメント活動を行い、特に製品の設計・開発にあたっては、リスクマネジメン トから得られた情報を加えることが必要である。つまり、市場からの製品(類似製品を含む)におけるフィ ードバック情報も重要な要素となる。 この活動はPDCAを基本としている。必ず市場からのサーベイ、顧客からの苦情、そういうものをフィー ドバックしてリスク分析に戻さないといけないのである。ここでいうリスクとは、「患者やユーザに対する健 康被害」のことである。「医療機器に欠陥が潜んでしまう」や「製造担当者がけがをする」といった事象は、 リスクマネジメントの対象ではないことに注意が必要である。 1つの医療機器の設計の全ライフサイクルを通じて、医療機器企業がリスクコントロールを最大限実施し たことを証明するために、リスクマネジメントにおける活動は、リスクマネジメントファイルにおいて、完 全に文書化しなければならない。 適切なリスクマネジメント活動は、新しい医療機器の開発、生産、販売等を支えることにより、重要な価 値をもたらす。 リスクマネジメント活動により、製品開発スケジュールへの影響を最小限に留めながら、機能的安全性や 使い勝手を向上させるための変更や改良の特定や実施が可能になる。 また、製品を市場に投入するまでの時間を短縮して、一層の競争優位性をもたらす。 2. 品質が良い医療機器とは 品質が良いということは、どんな状態であろうか。 医療機器においては、ユーザの要求事項と完成した医療機器の仕様が完全に適合している状態が、品質が 良いといえる。 使い勝手が良いとか、見てくれがいいとか、デザイン性がいいとか、そういうことは無関係なのである。 医療機器は、意図される使用目的(intended use)に対して機器が完全に適合している状態、これが、品質 が良い医療機器なのである。 要求事項と完成医療機器が完全に整合していることを確認して保証することを「バリデーション」と呼 ぶ。日本語では「妥当性の確認」と訳されている。 ISO-9000による定義には、「バリデーションとは、特定の意図した用途またはアプリケーションに関する要 求が満たされていることを、客観的な証拠の提示により確認することである。」と記載がる。 また、QSRには、「バリデーションとは、特定の意図する用途のための特有の要求事項が恒常的に満たされ 得るという客観的証拠の検討と規格による確認を意味する。」と記載されている。 バリデーションは、必ずユーザ環境またはユーザ環境を模擬した環境で実施しなければならない。医療機 器企業側のみで実施すると、実際の使用状況と異なり、想定外の事故が起きてしまう可能性があるのである。 ところが、医療機器には1つ問題がある。そもそも要求事項が正しいとは限らないのである。例えば、医 師などから、新規医療機器に対する要求が得られたとしよう。その要求に従い、設計し、製造したとしよう。 しかしながら、実際に使用してみた場合、実は当初想定したような使用目的にそぐわず、使いづらいなどの 問題がしばしば発生する。そのため、何らかのリスクが潜んでしまったり、時には事故が発生してしまうのである。 品質には4つの種類がある。 1. 企画の品質 2. 設計の品質 3. 製造の品質 4. サービスの品質 である。 それぞれ、品質の4つの種類における要求事項は、次の通りである。 企画の品質においては、製品で実現しようとしている特性に対する顧客の要求である。 設計の品質においては、企画の段階で検討された特性の水準や品質仕様である。 製造の品質においては、図面・仕様書などの設計文書である。 サービスの品質においては、調整、据え付け、消耗品の補給、不良品などへの対応に対する顧客の要求で ある。 これら要求事項を満たした場合、当該医療機器および付帯サービスは、品質が良いといえるのである。 3. ISO-13485の認証とFDA査察の違い ISO-13485は、「医療機器の品質マネジメントシステム−規制目的のための要求事項」である。 ISO-9001は、製造業からサービス業まであらゆる業種に対応可能な反面、医療機器に適用しようとす ると、その安全性を確保しきれないという問題があった。そこで制定されたのが、ISO-13485である。 ISO-9001をベースに、医療機器の安全性や品質を確保するのに必要な要求事項が追加された。 ISOに参加する各国は、国民の健康維持・向上のため、医療機器を厳しく規制している。ISO-13485が 「規制目的のための要求事項」とされているのは、このためである。 ISO-13485の目的は、世界中の医療機器法令の整合化を促進することである。つまり、ISO-9000は民 間の規格であるのに対して、ISO-13485はもはや規制要件なのである。 日本、米国、欧州など、ISOに参加する各国の当局がレビュを行っている。 ところで、ISO-13485の認証を受けていても、FDA査察で指摘されることがしばしばある。 ISO-13485の認証は、第三者認証機関(NB:Notified Body)が実施する。つまり民間業者がビジネス として実施するのである。クライアントとの付き合いもあり、必然的に厳しい指摘には及ばないことや 多くの指摘を出さないこともしばしばである。 しかしながら、FDAは米国民を保護するために、日本までやってきて査察を行うのであるから、リス クを徹底的に調査し、指摘を出すのである。査察官自身も国民への説明責任があり、また事故などが発生 した場合には、査察した者の責任が問われることもあり得るのである。 4. 放射線治療装置の死亡事故 1985年から1987年にかけて、米国で複数の医療施設でTherac-25という放射線治療装置が誤作動し、過大 な放射線を浴びた患者に死傷者が出た。 Therac-25という放射線治療装置は、旧モデルのTherac-20の廉価版であった。そのコストの下げ方は、電 気機械式の安全保護装置を、「安全はソフトウェアでも確立できる」というコンセプトのもと、経済性を優先 させるためにハードウェアによる安全装置を外した。そのためハードウェアによる安全装置のために抑えら れていたソフトウェアの不具合が発現した。 問題は、医療機器企業でのテストは、テストスクリプトに従ってテストデータを入力し、確認をしながら ステップを踏む。しかしながら、実際の医療現場では、医師がいわゆるブラインドタッチを行い、素早くデ ータを打ち込むことが多い。そのため、高エネルギーの放射線を照射してしまったために、被爆した患者が 数日後に亡くなったということであった。Therac-20では、ハードウェアによる安全装置が存在したため、当 該事故は発生せず、ソフトウェアの問題は露見しなかったのである。 当初は、調査したFDAも当該医療機器企業も、ハードウェアの故障と考えていた。そのため、なぜ過度な エネルギー照射が起こるのか原因を突き止めるのに長い時間がかかり、付け焼き刃の対策で装置の使用を止 めなかったために、Therac-25による犠牲者は6名にまで拡大してしまった。 この事故を調査した現マサチューセッツ工科大学のNancy Leveson教授は、事故調査を終えて次のような 教訓を書き残している。 ・ソフトウェアはどんなに慎重に作っても満足できるほどに完璧にはならないため、ソフトウェアがどん な動きをしたときでも、人命に危険が及ばないようにシステムを設計する必要がある(フェールセーフ、 独立した安全装置の設置など) ・ソフトウェアの信頼性をできるだけ高めるのは悪いことではないが、それでソフトウェアが安全になる わけではない。プログラマは要求を満たすようにコードを書くが、要求が正しいとは限らない。事故の ほとんどは、プログラミングのエラーではなく、要求が間違っていたり、不完全であったりしたために 起きている ・われわれの目標は、だれもがこの経験を生かせるようにすることであり、装置のメーカーや他の人を批 判することではない。間違いは、このメーカーにだけ起きたのではなく、残念ながら、安全が最優先さ れるべきその他のシステムにも、きわめて日常的に起きている ・いくつかの出来事が複雑に絡み合って起きた事故が、1つの原因にされることが多すぎる。ほとんどの 事故は、システム事故、つまり、さまざまな構成部分や機能が複雑に作用しあって起きるものだ。事故 の原因を1つに特定することは、大きな間違いだ。 Nancy Leveson 教授が語ったTherac-25の事故調査から導き出された組込みソフトウェアの安全に関する教 訓は、20年以上経過した現在でも生き続けている。 というよりも、組込みソフトウェアの大規模・複雑化とCPUの統合化により、より危険性が加速されてき おり、組込みソフトエンジニアがソフトウェア開発を行っている際に常に考えていなければいけない重要な 教訓となっている。 この事故を教訓に、FDAは1987年に「General Principles of Software Validation(ソフトウェアバリデーショ ンの原則)」と呼ばれるガイドラインを発行した。 ソフトウェアのバリテーションは、医療機器メーカーだけではなく、ユーザの環境またはユーザ環境を模 擬した環境で、ユーザが参画して実施することが義務付けられたのである。5. 誤使用 5.1 合理的に予見可能な誤使用 市場出荷された医療機器自体になんら欠陥がないとしよう。ところが、使う側であるユーザが操作を間違ってしまうということがあるのである。ヒューマンエラー、誤使用、不注意というものに対しても、必ず発生するので生産者側では気をつけなくてはならない。 医療機器の場合においても、こういった誤使用という問題がある。医療機器業者が想定した使用法が記載 されている取扱説明書の通りに使用しないというのが誤使用である。これはヒューマンエラーである。 いくら医療機器企業側が気をつけていても、ユーザ側が間違ってしまうということも、リスクとして捉え ていないといけないのである。 ISO/IEC Guide 51では、設計者等が想定した使用法(intended use)のみならず、「合理的に予見可能な誤使 用」(reasonably foreseeable misuse)も含んだ範囲でリスクを評価し、受け入れ可能なレベルに達するまで、リ スク低減を行う反復プロセスを図示している。(図2参照) 「合理的に予見可能な誤使用」をリスクアセスメントの範囲とすることを明記したことが極めて重要である。 5.2 誤使用の例 医療機器以外での誤使用、不注意に分類される事故例としては、ガスコンロの消し忘れで火がついてしまった、少ない油で天ぷらを揚げて火災になった、換気扇を回さずにガス湯沸かし器を使って一酸化炭素中毒になってしまったなどがあり得る。 リビングや寝室では、石油ストーブに洗濯物が落下して火災が発生した、付近の可燃物が発火した。湯た んぽでは低温やけどが発生した、電池を逆に入れてしまって破裂した、間違ってボタン電池を飲み込んでし まったなどが実際に起きている。 このようなことを含め、生産者はあらたな誤使用というリスクを発見した場合、そのリスクを回避するよ うな設計に変更にしなければならないのである。 または、誤使用を未然に防止するよう、ラベルなどに注意書きを記載しておかなければならない。例え ば、レーザーポインターには、「直接、目でのぞかないでください。」と注意書きが貼ってある。または携帯 用のカイロでは「肌に直接貼らないで下さい。低温やけどの恐れがあります。」と記載がある。 それでも、人は必ず間違いを起こすのである。 1980年の日航ジャンボ機の墜落事故は、ボーイング社の修理ミスだといわれている。医療事故では輸血す るときに血液型を間違えたとか、患者を取り違えたとか、薬品を間違った、手術のときに左右の肺を間違え た、そういったこともしばしば発生している。 株の大量誤発注という事件もあった。61万株を1株1円で売りに出してしまった、数量と金額を逆に間違 えて入力してしまったのが原因である。その際、東証のコンピュータには、発注取り消しという機能が設計 されておらず、被害が拡大した。 6. ISO-14971 6.1 ISO-14971とは ISO-14971「医療機器 - リスクマネジメントの医療機器への適用」は、医療機器企業が体外診断用医療機 器を含む医療機器に関連するハザードを特定し、リスクの推定および評価を行い、これらのリスクをコント ロールし、そのコントロールの有効性を監視する手順について規定しているというものである。 ISO-14971は、リスクマネジメントの原則と実践について示したものである。 ISO-14971は、医療機器に関する幾つかの主要な規格において参照されている。例えば、ISO-13485(医療 機器の品質マネジメントシステム)、IEC 60601-1(電気安全性)、IEC/EN 62366(医療機器の使い易さ)、ISO 10993(生物学的評価)、IEC62304(医療機器のソフトウェア)などである。 ISO-14971で示されたリスクマネジメントのプロセスは、次のような内容となっている。 6.2 ISO-14971の適用範囲 ISO-14971は、組織の営業活動から設計開発、製品製造、検査測定、製品納入、付帯サービスおよび市販後 の調査に至るすべてのライフサイクル活動に適用される。 しかしながら、大事なのは、医療機器の「設計・開発」プロセスにおいて、リスクマネジメント活動の結 果を取り入れて活動しなければならないということである。医療機器は医薬品と異なり、製造工程よりはむ しろ設計工程でリスクマネジメントを主に実施しなければならない。 設計・開発が適用除外される医療機器であっても、製品実現全体をとおしてリスクマネジメントのための 文書化された要求事項を確立することが要求されている。 リスクマネジメントの活動対象範囲は、組織が製造する製品すべてが対象となる。少量多品種であっても その一部を適用除外することは出来ない。また、付属品等も適用対象になる。 ただしISO-14971は、臨床的判断には適用しない。また、受容可能なリスクレベルを規定していない。 6.3 ISO-14971の経緯 ISO-14971の経緯を紹介する。(図3.参照)6.4 ISO-14971の構成 ISO-14971の目次を図4に記載する。 6.5 IEC-60601-1とISO-14971 IEC-60601-1には、「ISO-14971に適合するリスクマネジメントプロセスを実施する。」という要求事項があ る。また、要求事項の適合性の確認について、「適合性は、リスクマネジメントファイルを調査して確認す る」と記載がある。 しかし、IEC-60601-1の要求事項に適合していれば、その要求事項についてはリスクマネジメントを実施し なくても良いという「基礎安全」の考え方を取り入れている。 つまり、機器全体はリスクマネジメントISO-14971の管理下にあるが、その中でIEC60601-1の要求事項に よる部分があるということである。 IEC-60601-1の要求事項では、副通則であるEMC(IEC60601-1-2)、alarm(IEC60601-1-8)、usability (IEC60601-1-6)も含まれている。 7. EN ISO-14971 2012年7月31日、EN ISO-14971: 2009は、EN ISO-14971:2012に置き換わり、2009年版は2012年8月30 日の時点で廃止された。 EU地域では、EN ISO-14971:2012は、2013年1月より強制になった。EU圏へ輸出する製品への影響を評 価する必要がある。各国・地域の対応は、その国・地域の事情もあって独自路線をとるのはいたしかたない ところである。 EN ISO-14971:2012は、ISO-14971:2007の規定についての相違はないが、informative(参考)である附属書 Zs(Za、Zb、Zc)が変更されている。3つのEU医療機器指令(MDD:Medical Device Directive)の基本要件 (ER:Essential Requirements)の一部を加えたものになっている。 EN ISO-14971:2012は、以下の7つの点において、ISO-14971:2007と異なっている。 7.1.1 “negligible risk”(無視可能なリスク)の扱い “negligible risk”(無視可能なリスク)の扱いとして、ISO-14971の定義では“negligible risk”(無視可能な リスク)を放棄することができる場合があると記載されている。 EN ISO-14971では、全てのリスクについて、リスクマネジメントを実施しなければならないと記載され た。 7.1.2 リスクの受容可能性についての医療機器企業の自由裁量権 ISO-14971では、リスクの受容可能性の基準は医療機器企業が決定することになっている。その上で、受容 不可能なリスクに対してのみ、それらを統合した全体のリスク/便益分析を行うこととしている。 EN ISO-14971では、医療機器企業がリスクの受容可能性を判定するのではなく、全てのリスクは可能な限 り低減されなければならないとしている。 EN ISO-14971では、もはや受容可能性という概念はないに等しく、全てのリスクは他のリスクと組み合わ せてリスク/便益分析を実施しなければならない。 7.1.3 ALARPではなくAFAPによるリスク低減 ALARP(as low as reasonably possible:合理的に実行可能まで低く)ではなく、AFAP(as far as possible:可 能な限り)によるリスク低減を要求している。 ISO-14971では、リスクは「合理的に実行可能な程度まで(ALARP)」低減することと記載されてい るが、これは経済性を考慮した要求である。あまりにも規制を厳しくすると、コンプライアンスコストが 上昇し、製品の価格に上乗せされ、結果的には患者負担になってしまう。 EN ISO-14971では、「可能な限り(AFAP)」低減することを要求しているのである。 リスクを徹底的に低減するということは、開発コストがかかってしまい、経済性を無視しているという ことである。 7.1.4 あらゆるリスクに対するリスク/便益分析の適用 ISO-14971では、受容可能性を超えるリスクに対して、リスク/便益分析を要求している。 EN ISO-14971では、個々のリスクおよびすべての残存リスクに対するリスク/便益分析を要求してい る。 7.1.5 リスクコントロール手段についての裁量 ISO-14971では、下記3つのオプションから1つを選んでリスクコントロールを実施することを要求 している。 7.1.6 初期リスクコントロール手段についての裁量 ISO-14971では、設計による固有の安全性を要求している。 これに対し、EN ISO-14971では、可能な限りリスクを低減させることを要求している。設計および材 料・組立による安全性を要求しており、範囲が広い。 7.1.7 残存リスクについてのユーザへの情報提供 ISO-14971では、残存リスクについてのユーザへの情報提供は、リスクコントロール手段の1つであ るとしている。取扱説明書に記載するというのも、リスクコントロールの1つなのである。 EN ISO-14971では、残存リスクについてのユーザへの情報提供はリスク低減手段ではないとしてい る。 8. リスクマネジメント手順 本章では、設計開発のインプットとなるリスク分析、リスクを低減するためのリスクコントロール、リス クの受容可能性の決定とリスク/便益分析、また製造中および製造後の情報によるリスクマネジメントの見直 しといった、医療機器のリスクマネジメント手順について解説をする。 リスクマネジメント手順は、以下のステップにより構成される。 8.1 意図する使用および医療機器の安全に関する特質/医療機器の使用に関する特性の明確化(Step-1) このステップでは、対象となる医療機器について、意図する使用および合理的に予見できる誤使用を明確化する。 必要な場合には、安全に影響する定性的および定量的特性または必要な場合には限界を明確にする。 ユーザビリティに関係するハザード抽出のため、 8.2 ハザードの特定(Step-2) このステップでは、当該医療機器に対する既知のハザードおよび予見できるハザードを特定する。(図6参照) 8.3 リスクの推定(Step-3) Step-2で特定した「危害」つまりリスクに対して、「発生確率」および「重大性」を推定する。 いずれも定量的な表現か、または定性的な表現が用いられる。(図7参照) 8.4 リスクの評価(Step-4) Step-3で求めた発生確率と重大性から、リスクレベルを算出する。 リスクレベル=重大性×発生確率であるので、図7に従って、A3、A2、A1、B3、B2、B1、Cのいずれかになる。 リスクマネジメントワークシートに記載する際には、A、B、Cのいずれかで良い。 8.5 リスクコントロール Step-4で求めたリスクレベルにより、リスクコントロールの要・不要(リスクの許容可能性)を判定する。 リスクレベルがB以上の危険状態については、以下を実施する。 8.5.1 リスクコントロール手段の明確化(Step-5) リスクレベルの低減のための手段(リスクコントロール手段)を明確化する。 リスクコントロール手段としては、次の優先順位に従い、それらの1つ以上を使用すること。 8.5.2 IEC-62304によるソフトウェアに関する分析(Step-6) ソフトウェアを搭載した医療機器(単体プログラムを含む)の場合、IEC-62304の要求に従って、ソフトウ ェアの設計を実施しなければならない。 一般にソフトウェアの設計では、ソフトウェア要求仕様書(Software Requirements Specifications:SRS)に 従って、アーキテクチャ設計書を作成する。 アーキテクチャ設計書は、ソフトウェア要求仕様書に対応するソフトウェアの機能を記述し、さらに各機 能をソフトウェアアイテムに分割する。ソフトウェアアーキテクチャ設計の終了は、開発における最も初期 のポイントである。ソフトウェアアイテムの明確化により、安全性との関連が特定可能になる。 IEC-62304では、アーキテクチャ設計において分割されたソフトウェアアイテム毎に、ソフトウェアアイテ ムが寄与因子であり得るハザードから生じる、患者、オペレータまたはその他の人々に対する考えられる影 響により、ソフトウェア安全クラスを割り当てることを要求している。 ソフトウェア安全クラスは、危害の程度に基づいて次のいずれかに分類する。 クラスA:傷害が発生しない クラスB:深刻な傷害が発生しない クラスC:死亡または深刻な傷害が発生するおそれがある ただし、ハザードから生じる危険が、ハードウェアのリスクコントロールにより受容可能なレベルに低減 できるのであれば、それらを考慮に入れても良いとしている。 8.5.3 リスクコントロール手段の実施と文書化(Step-7) 選択したリスクコントロール手段を実施する。つまり、ハードウェアやソフトウェアによる安全装置の設計や、取扱説明書やラベルなどに使用女王の注意などを記載する等である。 実施したリスクコントロール手段は、それぞれの設計書等に文書化すること。 また、リスクコントロール手段が適切で有効であることを検証した記録を作成すること。 8.6 残留リスク評価(Step-8) リスクコントロール実施後に残留するリスクレベルを評価する。リスクコントロール実施により減少した発生確率および重大性を求め、リスクレベルを算出する。 リスクレベル=重大性×発生確率 リスククラスが、図7においてCになっていれば、受容可能と判断する。 受容可能であると判断した残留リスクについては、どの情報を提供(開示)するか、かつ、どの附属文書に記載するかを決定すること。 リスクレベルが受容可能なまで低減されていない場合は、Step-5に戻り、リスクコントロール手段を明確化すること。 特定した全ての危険状態からのリスクが考慮されていること(リスクコントロールの完全性)を明確にすること。 8.7 リスク/便益分析(Step-9) Step-8において、受容可能でないと判断し、かつ、それ以上のリスクコントロールの実施が現実的でない場合、データおよび文献を収集し、かつ、見直した上で、意図する使用の医学的効用が残留リスクを上回るかどうか判断する。 重大性が「重大」、「中程度」「軽微」と判定されたリスクであっても、リスク/便益分析を実施すること。 ただし、重大性が「致命的」なリスクが残留した場合は、その製品の開発を打ち切らなければならない。 8.8 新たなハザードの発生確認(Step-10) リスクコントロール手段の実施によって、新たなハザードが発生し、影響が出ていないかを調査する。以 下を考慮すること。 8.9 リスク評価の完了(Step-11) 上記のすべてのステップを踏み、リスクマネジメント活動が適切かつ完全に文書化されていることを確認した上で、個々の危険状態に対する一連のリスク評価活動が終了する。 完了した日付を、リスクマネジメントワークシートに記載すること。 8.10 残留リスクの全体的評価(Step-12) すべての危険状態に対して、残留リスクの全体的な評価を行う。結論としては、当該機器における残留リスクが受容できるか、受容できないかのいずれかとなる。 ここで、重大性が「致命的」なリスクがあってはならない。 また、残留リスクのリスククラスがB以上のものに対して、医学的効用がリスクを上回るものであることの判定が、根拠をもって実施されていなければならない。 8.11 製造中および製造後の情報によるリスクマネジメントの見直し手順 製造中および製造後に「新たな問題」が発生していないかを調査する。新たな問題が発生している場合は、 リスクの再評価を行うこと。 次の検討を行う。 9. 体外診断用医療機器(IVD)のリスクマネジメントモデル 体外診断用医療機器(体外診断用医薬品を含む)は、患者に直接接触することはない。つまり、当該体外診断用医療機器の欠陥により、患者がけがをすることはない。そのため、リスクの評価があいまいになりがちである。 図8に、体外診断用医療機器のリスクマネジメントモデルを示す。